Introduction
Microphysiological systems (MPS) are fast becoming a cornerstone of FDA-aligned non-clinical strategy, especially when paired with advanced in silico tools. The FDA Modernization Act 2.0 formally recognized non-clinical in vitro, in silico and in chemico methods as acceptable components of safety and efficacy packages, while Modernization Act 3.0 pushes toward qualification frameworks and expedited reviews when new-approach methodologies (NAMs) are used appropriately. In this article, we explain how this initiative affects preclinical research on monoclonal antibodies and, within this framework, how a breast cancer organoid preclinical model for mAb development exemplifies how MPS can reduce animal use while delivering human-relevant evidence in one of pharma’s busiest therapeutic areas.
Learn more about our ready to use breast cancer organoid models.

FDA modernization act drives the use of MPS
The Food & Drug Administration marked 2022 with its FDA Modernization Act 2.0 formally recognizing some in vitro and in silico methods as acceptable components of IND/BLA packages in lieu of animal testing; The following Modernization Act 3.0 (Feb 2024) now directs the Agency to qualify nonclinical methods on an accelerated timeline and expedite review for applications using qualified NAMs. In this context, the industry guidance urges a regulatory “safe harbor” so sponsors can include hybrid packages (traditional + NAMs) while qualification matures, as shown in recent comments co-authored by actors from different pharmaceutical companies(1,2).
Building on that legislative foundation, the FDA’s Roadmap to Reducing Animal Testing in Preclinical Safety Studies puts monoclonal antibodies (mAb) at the front of the transition, citing species-specific pharmacology and immunology as key reasons to begin animal reduction with biologics before expanding to other possible molecules. A concrete example in this plan is the possibility to shorten routine primate chronic toxicology from six months to three for mAbs when a clean 1‑month study is supplemented by new approach methodologies (NAMs)(3).
Operationally, this is a signal from the FDA that sponsors may submit NAM data in parallel with animal data to build the shared evidence base. Acceptance will be formalized via Drug Development Tool (DDT) qualification, notably with the FDA’s Innovative Science and Technology Approaches for New Drugs (ISTAND) program which support the development of novel approaches to drug development that may be acceptable for regulatory use and therefore anchors each method to a defined context of use (COU) so that, once qualified, any sponsor can rely on it with predictable review.
For breast oncology programs, this translates into early use of MPS, including breast cancer organoid and breast cancer on chip platforms, paired with PBPK/QSP and AI/ML safety prediction to answer decision‑enabling questions more human‑relevantly than historical animal tests, while advancing toward reduced or waived studies under a weight‑of‑evidence framework. Early INTERACT meetings can align expectations and clarify data-acceptance criteria. Notably, defining fit‑for‑purpose metrics and SOPs up front will smooth IND‑enabling strategy and GLP‑adjacent rigor.
Why monoclonal antibodies lead, and how MPS bridge the gap
The decision to focus on mAbs is not random and makes sense both from a market and biological standpoints.
Firstly, since 1995 the share of biologics in the global drug‑development pipeline has risen from 15 % to 50 %(4). Much of that growth comes from oncology, where immuno‑oncology is dominating. Therefore it’s expected that regulators face an increasing surge of antibody‑based INDs that must be evaluated under the new animal‑reduction mandate.
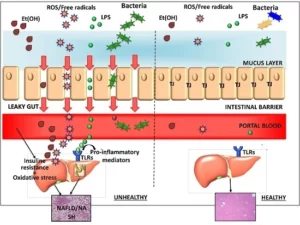
On the biological side, animal models answer only part of the question. For small‑molecule drugs, conserved enzyme targets and metabolic routes mean that animal pharmacokinetics and toxicity often translate. However it is different for biologics: their epitopes and Fcγ‑receptor interactions are uniquely human, FcRn binding varies by species, and treatment can trigger anti‑drug antibodies (ADA) that skew exposure and hide toxicity. The 2006 TGN1412 cytokine‑storm catastrophe, benign in monkeys but disastrous in human volunteers, notably highlighted this effect and drove routine use of in vitro human cytokine‑release assays (CRA) with PBMC or whole blood(5).
MPS are designed to generate human‑relevant datasets that close the translational gap left by animal studies. Ingber et al. 2022 reviewed a decade of organ‑on‑chip progress and concluded that properly validated chips can outperform traditional species across pharmacology and toxicology endpoints, a finding that is particularly relevant for large‑molecule therapeutics(6). A flagship example is the Human Liver‑Chip, which detected 87 % of drugs known to cause clinical hepatotoxicity, surpassing animal and 2D assays and now progressing through the FDA’s ISTAND pathway(7). Building on that benchmark, cross‑disciplinary teams are building liver, cardiac and tumor compartments into integrated circuits so that mAbs, bispecifics and ADC payloads can be profiled for on‑target efficacy and off‑target safety within human‑specific workflows. Concrete examples already exist: a lung‑alveolus chip predicted the cytokine‑driven lung inflammation provoked by a high‑affinity T‑cell bispecific antibody and correctly ranked a lower‑affinity variant as safer, while an endothelium‑lined vessel chip reproduced antibody‑induced thrombosis that traditional rodent studies failed to detect.
With the FDA inviting parallel NAM submissions and accelerating ISTAND/DDT qualification, developers who feature validated MPS early in their biologics pipeline can shorten timelines, document animal reduction and predict human outcomes with greater confidence.
As breast cancer is now the world’s second‑most targeted disease area and commands a substantial share of antibody‑based clinical trials, it makes an ideal therapeutic field to illustrate how mAb‑focused MPS can transform IND strategy. The next section explores those breast‑cancer‑specific platforms and data in detail.
Breast cancer organoids and chips: human-relevant data for mAb pipeline
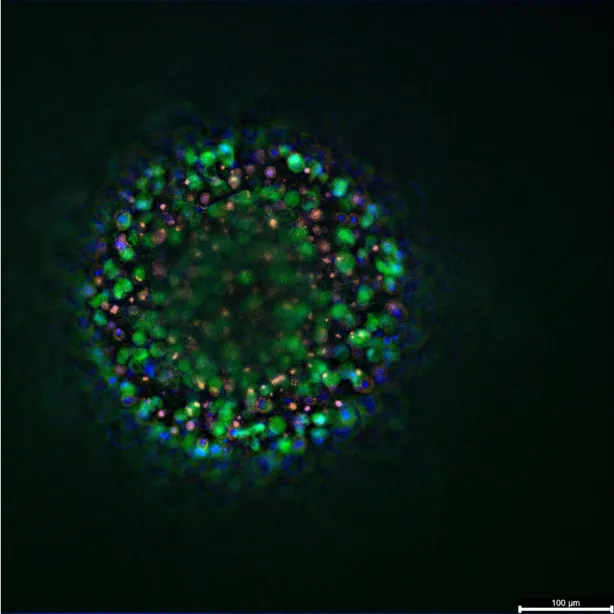
Breast cancer organoid biobanks now span every molecular subtype and, when merged with microfluidic perfusion, recreate the dynamic tumour–immune interplay targeted by immunotherapies. Guan et al. catalogue platforms that embed tumour epithelium, fibroblasts and autologous lymphocytes in 3D matrices, then quantify checkpoint‑blockade or cytokine responses with single‑cell resolution. These immune‑competent cultures supply the first human system in which mAb‑driven cytokine release, receptor occupancy and tumour‑cell lysis can be evaluated side‑by‑side(8).
Extending these insights into perfused, vascularised settings, Giannitelli et al. review breast‑tumour‑on‑chip technologies that integrate stroma, endothelium and immune cells under flow. They report oxygen, drug and shear gradients that mirror PBPK compartments, enabling direct measurement of antibody permeability and binding kinetics for QSP refinement and MABEL calculation. These can be then used for weight‑of‑evidence submissions(9).
Two recent studies illustrate how such chips translate to actionable decisions. Nguyen et al. reconstructed a HER2‑positive microenvironment on‑chip and showed that trastuzumab prolongs immune synapses and triggers antibody‑dependent cellular cytotoxicity (ADCC); cancer‑associated fibroblasts attenuated the response, revealing a stromal resistance node missed by animals(10). Building on that platform, Perucca et al. used the MIRO model to demonstrate that IL‑2 can breach the CAF barrier and restore anti‑HER2 ADCC, guiding rational cytokine‑mAb combination design before IND filing(11).
Collectively, these studies deliver a template for hybrid non‑clinical packages: 1) immune‑competent tumour chips for mechanistic potency and safety (CRA, ADCC, tissue cross‑reactivity), 2) liver‑ and cardiac‑chip add‑ons for systemic studies, and 3) translational modelling that links chip exposure to projected clinical PK/PD. The remaining barrier is consistency. The final section turns to the standardisation, benchmarking and qualification efforts that must mature before breast‑cancer MPS can become a routine IND asset.
Standardization for better adoption
MPS already generate data in a format regulators recognise, but the results must be shown to hold up in every laboratory. Achieving that consistency hinges on three practical steps that will turn MPS into qualified new‑approach methodologies (NAMs). Reference compound sets and cross‑lab ring trials, led by the IQ MPS Affiliate and NCATS Tissue‑Chip initiative, will anchor performance metrics and control limits. GLP‑aligned SOPs and digital audit trails must accompany every tumour‑chip run so that datasets can slot directly into eCTD modules. Finally, sponsors should engage ISTAND or DDT qualification early, defining a clear context of use and submitting paired animal‑plus‑chip data to create a regulatory “safe harbour.”
If sponsors meet these benchmarks, breast‑cancer organoid and on‑chip assays will shift from exploratory tools to accepted IND evidence. That change should reduce animal use, compress timelines to first‑in‑human trials, and improve the accuracy of clinical predictions.
Resources
- Stresser DM, Kopec AK, Hewitt P, Hardwick RN, Van Vleet TR, Mahalingaiah PKS, et al. Towards in vitro models for reducing or replacing the use of animals in drug testing. Nat Biomed Eng. 27 déc 2023;8(8):930‑5.
- Carratt SA, Zuch De Zafra CL, Oziolor E, Rana P, Vansell NR, Mangipudy R, et al. An industry perspective on the FDA Modernization Act 2.0/3.0: potential next steps for sponsors to reduce animal use in drug development. Toxicol Sci. 1 janv 2025;203(1):28‑34.
- FDA. Roadmap to Reducing Animal Testing in Preclinical Safety Studies [Internet]. Available at:: https://www.fda.gov/files/newsroom/published/roadmap_to_reducing_animal_testing_in_preclinical_safety_studies.pdf
- Lloyd I. Whitepaper: Pharma R&D 2025. Citeline.
- Suntharalingam Ganesh, Perry Meghan R., Ward Stephen, Brett Stephen J., Castello-Cortes Andrew, Brunner Michael D., et al. Cytokine Storm in a Phase 1 Trial of the Anti-CD28 Monoclonal Antibody TGN1412. N Engl J Med. 355(10):1018‑28.
- Ingber DE. Human organs-on-chips for disease modelling, drug development and personalized medicine. Nat Rev Genet. août 2022;23(8):467‑91.
- Ewart L, Apostolou A, Briggs SA, Carman CV, Chaff JT, Heng AR, et al. Performance assessment and economic analysis of a human Liver-Chip for predictive toxicology. Commun Med. 6 déc 2022;2(1):154.
- Guan D, Liu X, Shi Q, He B, Zheng C, Meng X. Breast cancer organoids and their applications for precision cancer immunotherapy. World J Surg Oncol. 26 oct 2023;21(1):343.
- Giannitelli SM, Peluzzi V, Raniolo S, Roscilli G, Trombetta M, Mozetic P, et al. On-chip recapitulation of the tumor microenvironment: A decade of progress. Biomaterials. avr 2024;306:122482.
- Nguyen M, De Ninno A, Mencattini A, Mermet-Meillon F, Fornabaio G, Evans SS, et al. Dissecting Effects of Anti-cancer Drugs and Cancer-Associated Fibroblasts by On-Chip Reconstitution of Immunocompetent Tumor Microenvironments. Cell Rep. déc 2018;25(13):3884-3893.e3.
- Perucca A, Llonín AG, Benach OM, Hallopeau C, Rivas EI, Linares J, et al. Micro Immune Response On-chip (MIRO) models the tumour-stroma interface for immunotherapy testing. Nat Commun. 3 févr 2025;16(1):1279.
FAQ
The Food & Drug Administration formally acknowledged certain non-animal test methods through the FDA Modernization Act 2.0. This act recognized that laboratory-based and computational methods are acceptable components of safety and efficacy packages, sometimes in place of animal testing. A subsequent act, Modernization Act 3.0, directs the agency to qualify nonclinical methods on a faster timeline. It also pushes for expedited reviews for applications that use qualified newer methods. Industry guidance suggests sponsors can include hybrid packages, which contain both traditional and newer test data, while the qualification process matures. This legislative foundation is driving a shift in how non-clinical strategy is approached.
Monoclonal antibodies (mAbs) are placed at the front of the transition in the FDA’s plan. The plan cites reasons related to pharmacology and immunology that are particular to different species. These reasons support beginning animal reduction with biologics before moving to other molecules. A concrete example is provided in the plan. The possibility exists to shorten routine primate chronic toxicology studies. A six-month study might be reduced to three months for antibodies. This reduction is allowed when a clean one-month study is supplemented by alternative test methods. This focus signals that sponsors may submit new method data alongside animal data.
Acceptance of newer testing methods will be formalised through a "Drug Development Tool" qualification process. This is supported by the FDA’s program for "Innovative Science and Technology Approaches for New Drugs". This program helps with the creation of new approaches to drug development. These approaches may be found acceptable for regulatory use. Each method is anchored to a defined "context of use". Once a method is qualified for that particular context, any sponsor can rely on it with predictable review. For breast oncology, this means early use of advanced cellular models, like breast cancer organoids. These are paired with computational safety predictions.
The decision to focus on antibodies is based on market and biological factors. First, the share of biologics in the global drug-development pipeline has risen greatly, from 15 per cent to 50 per cent since 1995\. Much of this growth is in oncology. Regulators are expected to face an increasing surge of antibody-based applications that must be evaluated under the new animal-reduction mandate. On the biological side, animal models provide only partial answers for biologics. Their epitopes and Fcγ-receptor interactions are uniquely human. FcRn binding also varies by species. These differences make animal data less translatable than for small-molecule drugs.
The TGN1412 incident in 2006 was a catastrophe. The drug was found to be benign in monkeys. It was disastrous in human volunteers, causing a cytokine storm. This event showed a major gap in testing. It demonstrated that animal models do not always predict human immune responses to biologics. The incident led to the routine use of laboratory-based human cytokine-release assays (CRA). These assays use peripheral blood mononuclear cells (PBMC) or whole blood. This event is a clear example of why animal data for biologics can be insufficient. It also showed why human-based laboratory systems are needed.
Advanced biological models are designed to generate human-relevant datasets. These datasets help close the gap left by animal studies. It has been concluded that properly validated microscale biological systems can outperform traditional species. This finding applies across pharmacology and toxicology endpoints. This is particularly true for large-molecule therapeutics. These systems can model human-specific interactions that animal models cannot. For example, they can be used to profile antibody payloads for on-target efficacy. Off-target safety within human-specific workflows can also be assessed. This provides a more accurate prediction of human outcomes.
A flagship example provided is a human liver micro-device. This microscale biological system was evaluated. It was found to detect 87 per cent of drugs known to cause clinical hepatotoxicity. This performance was better than animal and 2D assays. This model is now progressing through the FDA’s qualification pathway for innovative science approaches. Building on this standard, cross-disciplinary teams are now constructing integrated circuits. These circuits may contain liver, cardiac, and tumour compartments. This allows antibodies, bispecifics, and antibody-drug conjugate (ADC) payloads to be profiled for safety and efficacy in human-specific laboratory workflows.
Breast cancer organoid biobanks are available. They now cover every molecular subtype of the disease. When these organoids are merged with microfluidic perfusion, they can recreate the tumour–immune interplay. This interplay is the target of immunotherapies. Platforms have been catalogued that embed tumour epithelium, fibroblasts, and autologous lymphocytes in 3D matrices. These systems can then quantify responses to checkpoint blockade or cytokines with single-cell resolution. These immune-competent cultures supply the first human system. In this system, antibody-driven cytokine release, receptor occupancy, and tumour-cell lysis can all be evaluated side-by-side.
Perfused micro-devices for breast tumours integrate stroma, endothelium, and immune cells under flow. These systems are reported to have oxygen, drug, and shear gradients. These gradients are similar to physiological compartments. This allows for the direct measurement of antibody permeability. Binding kinetics can also be measured. These data can be used for refining computational models. They can also be used for weight-of-evidence submissions. For instance, a HER2-positive microenvironment was reconstructed on a micro-device. It showed that trastuzumab prolongs immune synapses and triggers antibody-dependent cellular cytotoxicity (ADCC). This showed a stromal resistance mechanism not seen in animals.
These advanced cellular models already generate data in a format that regulators can recognise. The results must be shown to hold up in every laboratory. Achieving this consistency depends on three practical steps. First, reference compound sets and cross-lab ring trials will anchor performance metrics. Second, rigorous procedural documentation and digital audit trails, similar to good laboratory practices, must accompany every perfused cellular model run. This ensures datasets can slot directly into regulatory modules. Finally, sponsors should engage with qualification programs early. A clear "context of use" should be defined. Paired animal-plus-chip data can be submitted. This creates a regulatory “safe harbour.”