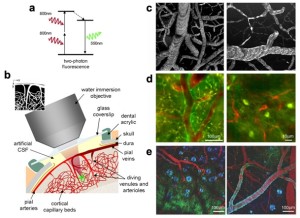
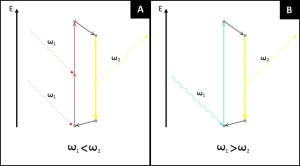
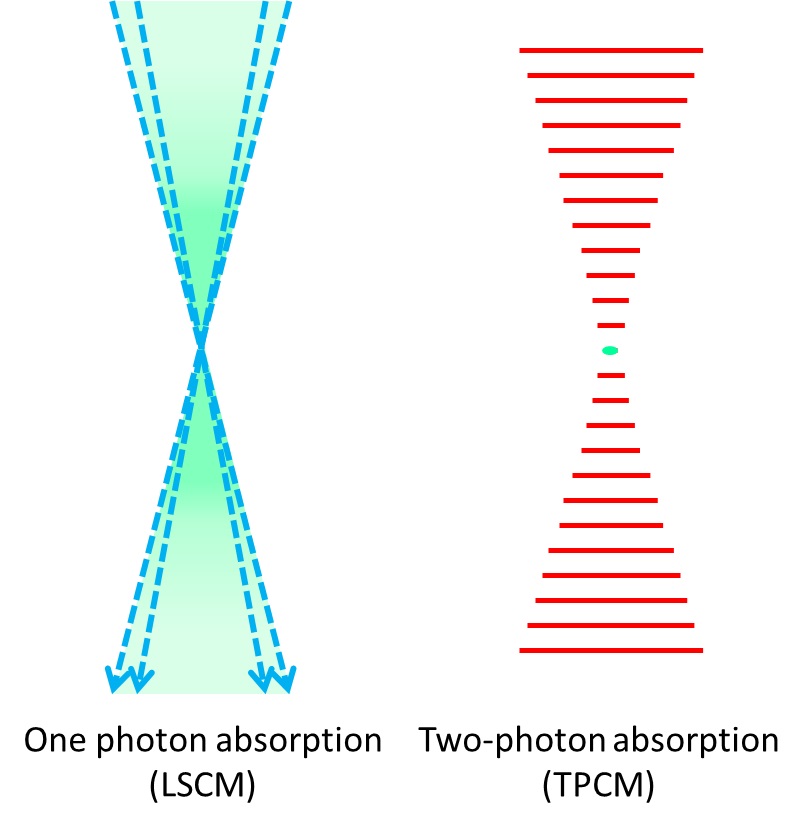
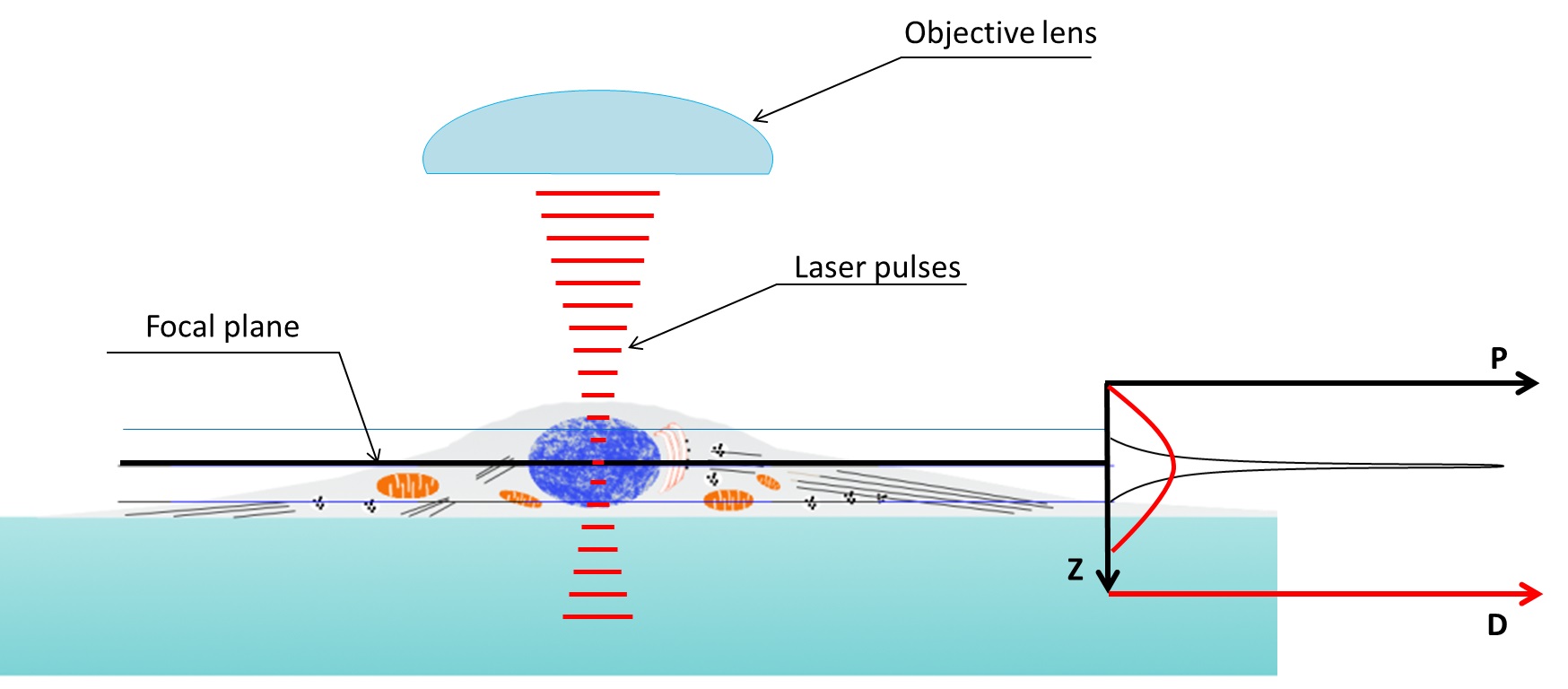
Two-photon excitation microscopy is a specific imaging technique. It is based on the capability, under certain conditions, to excite one electron from its ground state by using two photons. The light used to create the excitation has a longer wavelength than the fluorescent light that is collected. This is different from single-photon methods, where the excitation light has more energy than the emitted light. The principle was originally predicted by M. Goeppert-Mayer in 1931 and later demonstrated in 1961\. Because longer wavelengths are used, the method is considered cell-friendly. It allows for the imaging of living specimens with few, if any, physiological alterations.