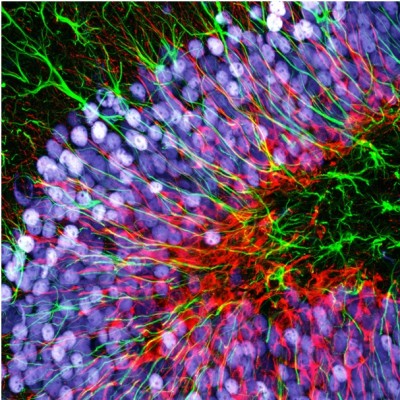
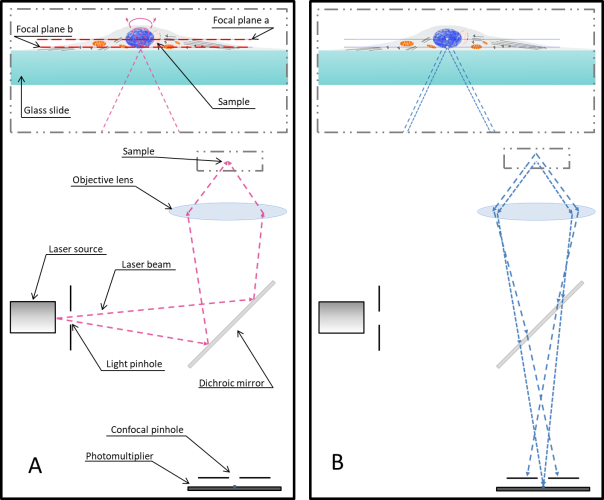
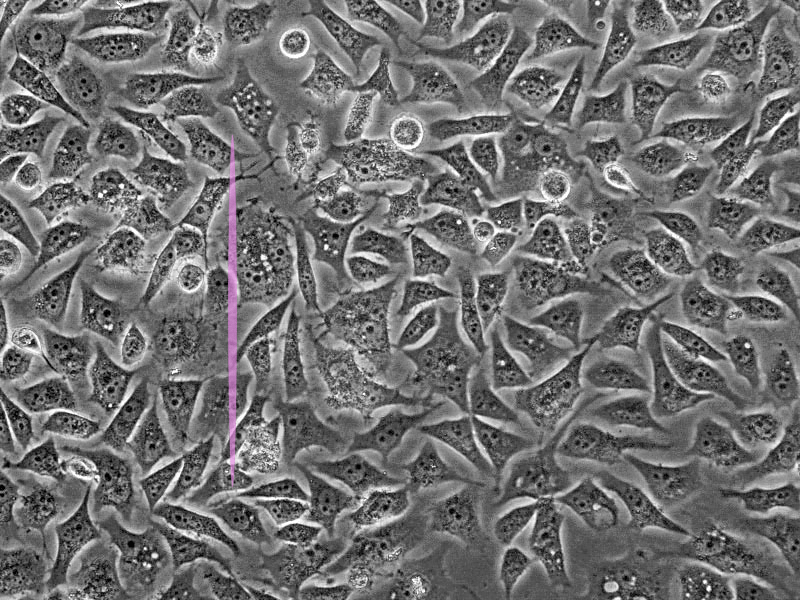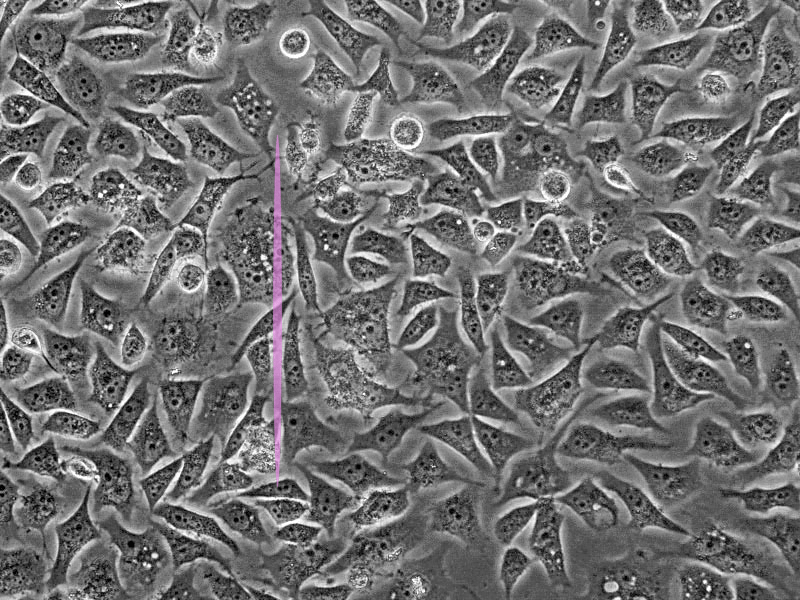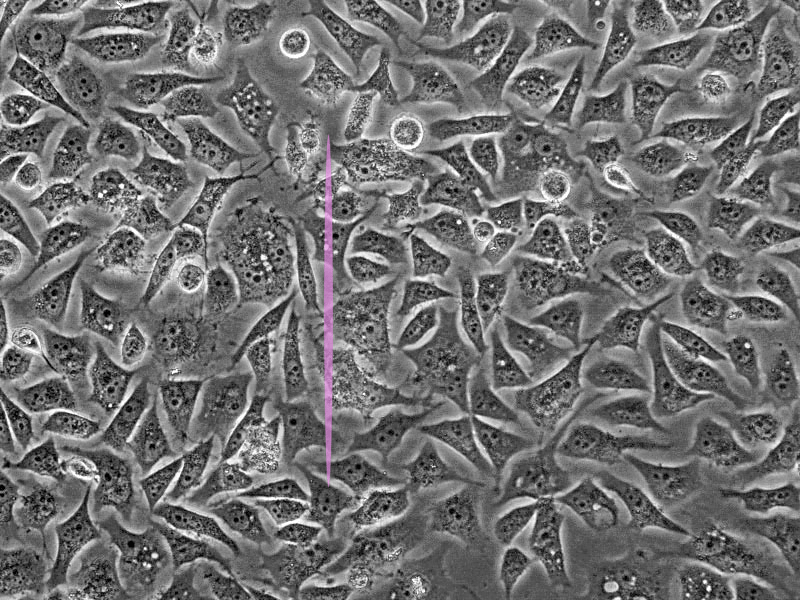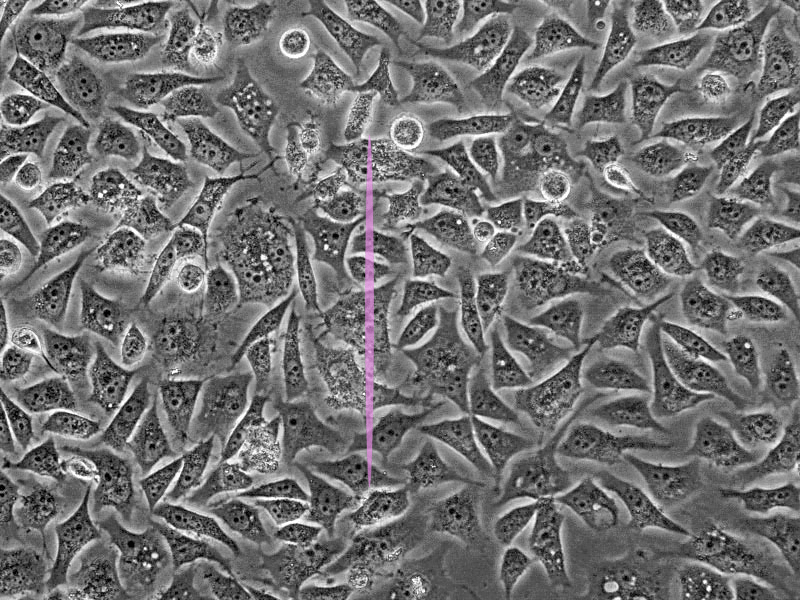
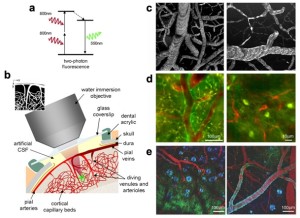
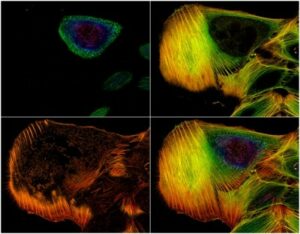
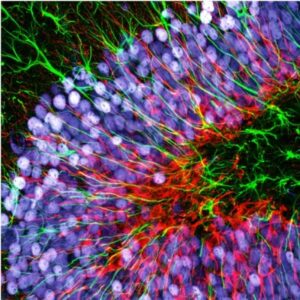
One of the main drawbacks is the scanning speed. The light is concentrated in a small spot, which must be moved at high speed to scan the entire sample. This process is slower than wide-field illumination. A second limitation is related to the confocal pinhole. While the pinhole’s selectivity is good for reducing noise, it also greatly decreases the amount of light that goes to the sensor. Therefore, the sensor must be very sensitive, or the exposure time must be increased. Highly sensitive sensors, such as photomultipliers, can be prone to generating false signals, or noise. This noise is generally removed by repeating the scan, but this action further increases the total scanning time.