Liver disease and drug-induced liver injury remain critical challenges in medicine and pharmaceutical development, demanding models that closely replicate human liver biology. In recent years, major progress has come from key studies on liver organoids and liver-on-chip systems, which better capture cellular complexity, metabolic function, and therapy responses than conventional cell lines or animal models. These approaches are increasingly supported by both academia and regulatory agencies as reliable, human-relevant platforms for understanding liver physiology, modeling disease, and predicting drug toxicity.
In this article, we highlight ten influential publications from 2024–2025 that showcase how organoid and organ-on-chip technologies are advancing liver research. Together, these studies demonstrate the potential of human-derived in vitro systems to transform drug discovery, reduce reliance on animal testing, and accelerate the development of safer, more effective therapies.
Learn more about our ready to use human liver organoid models.

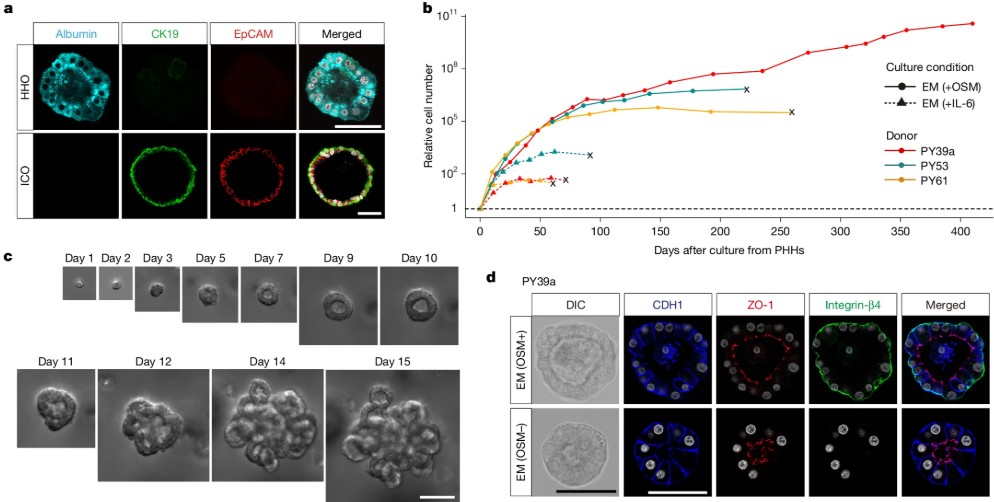
Human Adult Hepatocyte Organoids That Finally Grow and Stay Differentiated
For years, drug discovery has been held back by a simple trade-off: primary human hepatocytes keep adult functions but don’t expand, while expandable cultures drift into ductal metaplasia and lose liver identity. That’s bad news for modeling metabolism, DILI, and inherited disorders where you need weeks of stable, adult-like function.
In a 2025 Nature paper, Igarashi et al.(1) resolve that trade-off with a new human adult hepatocyte organoid system. Guided by cancer-genomics clues to niche signaling, they blend EGF/HGF/FGF10, Wnt/R-spondin, TGFβ inhibition, and, crucially, oncostatin M (OSM)–STAT3 activation to drive long-term self-renewal without flipping hepatocytes into a biliary fate. The organoids expand from single ASGPR1⁺EpCAM⁻ hepatocytes, maintain hepatic identity, and even repopulate mouse liver after transplantation, reinstating metabolic zonation markers in vivo.
When the team removes niche factors and adds a hormone cocktail (growth hormone, prolactin, cortisol) plus DAPT, with a “fasting” step or Wnt adjustment, they generate differentiated organoids that form bile canaliculi, secrete and conjugate bile acids, produce albumin and coagulation factors, and display CYP activities (including rifampicin-inducible CYP3A). Importantly for toxicology, the model shows clinically coherent acetaminophen injury at relevant exposures, and the damage is rescued by N-acetylcysteine, mirroring the emergency-room antidote. They also demonstrate clean gene-edited disease models (e.g., G6PC and OTC knockouts) that reproduce pathway deficits in gluconeogenesis and the urea cycle. Together, this is a rare organoid platform that’s simultaneously expandable and adult-functional; a practical step toward higher-fidelity liver organoid assays for drug discovery and mechanistic disease modeling.
Multi-Zonal Human Liver Organoids Capture Functional Zonation
One of the major limitations of in vitro liver models has been the absence of metabolic zonation, the spatial division of labor across periportal (zone 1) and pericentral (zone 3) hepatocytes. This spatial heterogeneity underlies critical functions such as nitrogen metabolism, bile acid processing, and zone-specific susceptibility to hepatotoxicants. Conventional liver organoid systems typically lose this organization, limiting their predictive value for both physiology and toxicology.
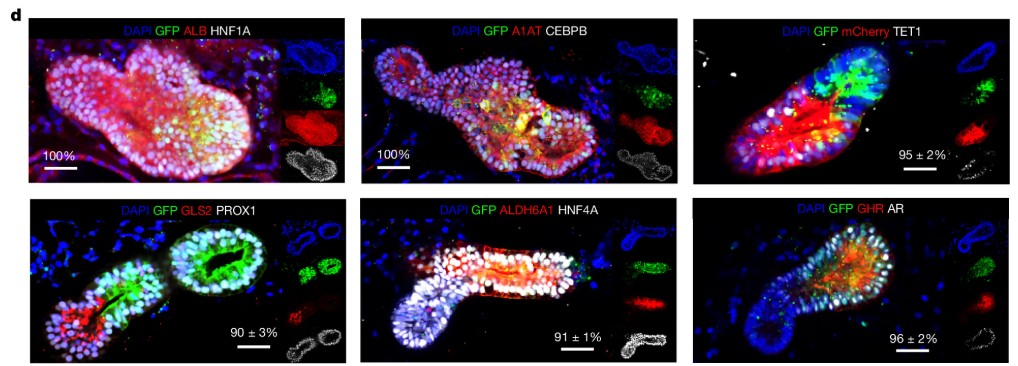
Reza et al. (Nature, 2025)(2) report the generation of multi-zonal human liver organoids (mZ-HLOs) from pluripotent stem cells. By engineering hepatic progenitors with inducible ascorbate synthesis to promote zone 1 identity and applying low-dose bilirubin to promote zone 3 fate, the authors created distinct zonal populations that self-assembled into fused organoids. Single-nucleus RNA sequencing confirmed the presence of periportal, interzonal, and pericentral hepatocyte clusters, together with cholangiocytes, endothelial cells, stellate cells, and macrophages. Chromatin profiling identified EP300 as a central regulator of zonal gene expression, cooperating with TET1 in zone 1 and HIF1A in zone 3.
Functionally, the organoids reproduced zone-specific responses to toxicants, with allyl alcohol selectively injuring periportal regions and acetaminophen affecting pericentral zones. Transplantation into bile-duct-ligated rats improved survival and restored key metabolic functions, including ammonia detoxification and albumin secretion. These findings demonstrate that multi-zonal liver organoids provide a physiologically relevant model for studying liver function, drug metabolism, and zone-specific hepatotoxicity in a human context.
Vascularized Organoids Model Early Human Cardiac and Hepatic Development
The formation of a functional vasculature is a defining feature of early organogenesis, yet this process is largely inaccessible for direct study in human embryos. Existing organoid systems often lack a structured vascular network, limiting their fidelity for modeling development and constraining their growth and viability.
Abilez et al. (Science, 2025)(3) addressed this limitation by combining micropatterned human pluripotent stem cells (hPSCs) with a systematic screening of differentiation conditions. The team established gastruloid-based models carrying four lineage-specific fluorescent reporters, enabling precise tracking of mesoderm, endoderm, and cardiovascular progenitor formation. By applying a defined cocktail of growth factors and small molecules, they generated cardiac vascularized organoids (cVOs) with a branched, lumenized vascular network integrated into multilineage cardiac tissue, including cardiomyocytes, endothelial cells, smooth muscle cells, and pericytes. Single-cell transcriptomics and high-resolution 3D microscopy confirmed that these organoids shared structural and functional features with a 6.5-week human embryonic heart.
Importantly, the same vascular-inducing strategy produced hepatic vascularized organoids (hVOs) that contained hepatocytes interwoven with endothelial networks. Functional analyses demonstrated that the vascular component was essential for organoid maturation and metabolic performance. The findings suggest that a conserved developmental program underlies vascularization across organs, and that this system can be used to investigate early human organ development as well as the effects of drug exposures on vascularized cardiac and hepatic tissues.
Encapsulated Human Hepatocyte Organoids for Liver Failure Therapy
Cell-based therapies for acute and chronic liver failure are limited by the scarcity of donor hepatocytes and their rapid loss of function after transplantation. Conventional primary hepatocyte grafts often fail to proliferate or integrate efficiently, restricting their clinical applicability.
Yuan et al. (Cell Stem Cell, 2024)(4) report the generation of encapsulated proliferating human hepatocyte organoids (eLOs) as a potential therapeutic alternative. The approach begins with proliferating human hepatocytes (ProliHHs), which were expanded in vitro and then matured into three-dimensional organoids. Encapsulation within alginate provided both structural support and immunoprotection, allowing the organoids to be transplanted intraperitoneally without host rejection.
In mouse models of post-hepatectomy liver failure and acetaminophen-induced acute liver injury, eLOs restored critical liver functions, including albumin production, ammonia detoxification, and glucose regulation. Treated animals showed improved survival and accelerated regeneration of native liver tissue. Mechanistic analysis further revealed that eLO transplantation enhanced intestinal barrier integrity and reduced systemic inflammation, thereby supporting overall recovery.
Comprehensive safety assessments demonstrated that encapsulated organoids were genetically stable, non-tumorigenic, and restricted to the implantation site. These findings establish eLOs as a promising platform for preclinical liver cell therapy, combining scalability, safety, and durable hepatic function in a clinically relevant organoid format.
Self-Organization of Sinusoidal Vessels in Human Liver Bud Organoids
Reproducing organ-specific vasculature remains a critical challenge in organoid research. In the liver, sinusoidal endothelial cells (LSECs) are essential for hepatocyte maturation, nutrient exchange, and coagulation factor production. Most existing vascularized liver organoid systems lack bona fide sinusoidal features, limiting their utility for modeling physiology and disease.
Saiki et al. (Nature Biomedical Engineering, 2024)(5) established a strategy to generate human liver bud organoids (HLBOs) with self-organized sinusoidal networks. By directing pluripotent stem cells into liver sinusoidal endothelial progenitors (iLSEPs), then combining them with hepatic endoderm, mesenchyme, and arterial progenitors in a multilayered air–liquid interface (IMALI) culture, the authors obtained organoids that contained hepatocyte clusters neighbored by multiple endothelial subtypes. The resulting vasculature included LYVE1⁺STAB1⁺CD32b⁺ sinusoidal-like cells with fenestrae, as well as central vein–like endothelial populations.
Functional analyses showed that WNT2-mediated angiocrine signaling from sinusoidal cells enhanced hepatocyte differentiation and metabolic activity. Intravital imaging of transplanted HLBOs in mice confirmed the formation of fully perfused human vessels with sinusoid-like permeability. Critically, the organoids secreted coagulation factors (V, VIII, IX, XI) sufficient to correct clotting defects in human plasma assays and to rescue the bleeding phenotype in hemophilia A mouse models.
This work demonstrates that sinusoidal self-organization within liver organoids is achievable, and that these systems can reproduce both endothelial specialization and functional liver support, positioning them as advanced models for vascularized liver biology and for preclinical studies of coagulation disorders.
Epithelial Plasticity in Chronic Liver Disease
The capacity for regeneration in the human liver during chronic injury has remained uncertain, as most evidence has come from animal models. Whether hepatocytes and cholangiocytes in diseased human livers can undergo lineage plasticity has been a long-standing question.
Gribben et al. (Nature, 2024)(6) addressed this by combining single-nucleus RNA sequencing (snRNA-seq) of 47 patient liver biopsies across the spectrum of metabolic dysfunction–associated steatotic liver disease (MASLD) with advanced three-dimensional imaging. Their analyses revealed progressive architectural disruption: hepatocytes lost zonation, the biliary tree underwent extensive remodeling, and populations of cells emerged that co-expressed hepatocyte and cholangiocyte markers. These biphenotypic cells increased with disease severity, indicating that epithelial plasticity is an acquired feature of chronic injury.
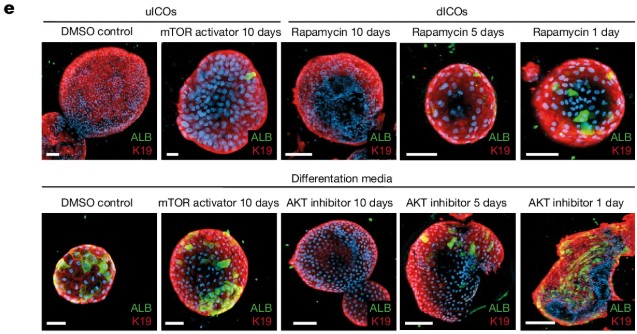
To test the mechanisms underlying this process, the authors generated intrahepatic cholangiocyte organoids (ICOs) from end-stage MASLD livers. When differentiated in vitro, these organoids recapitulated cholangiocyte-to-hepatocyte transdifferentiation, validating the observations in patient tissue. Functional studies identified the PI3K–AKT–mTOR pathway as a critical regulator of this plasticity, linking it directly to insulin signaling and metabolic dysfunction.
These findings demonstrate that chronic human liver disease is associated with a profound gain of epithelial plasticity, in which hepatocytes and cholangiocytes adopt hybrid identities in the absence of classical stem cell activation. The work provides mechanistic insight into the pathology of MASLD and establishes patient-derived organoids as a tractable system to model transdifferentiation and test potential therapeutic interventions targeting plasticity pathways.
Pharmacogenomic Profiling of Liver Cancer Organoids
Inter- and intra-tumor heterogeneity is a central challenge in liver cancer, undermining the effectiveness of targeted therapies. Clinical responses to multikinase inhibitors such as sorafenib and lenvatinib vary widely, and predictive biomarkers are lacking.
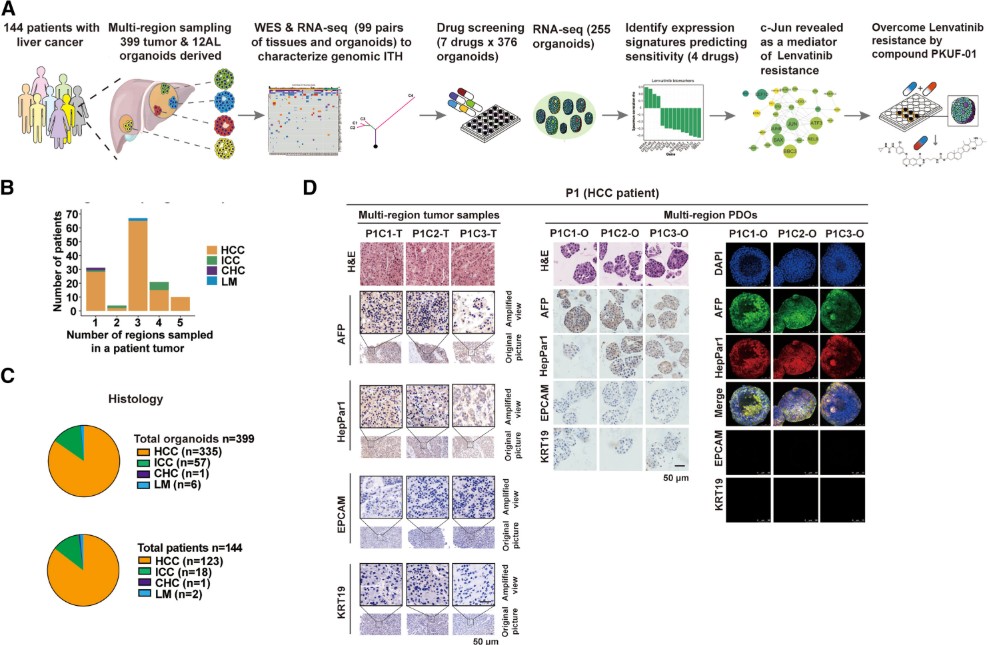
Yang et al. (Cancer Cell, 2024)(7) established a biobank of 399 patient-derived liver cancer organoids from 144 patients, including hepatocellular carcinoma, intrahepatic cholangiocarcinoma, and mixed subtypes. These organoids faithfully recapitulated the histology, genomic landscape, and transcriptomic signatures of their parental tumors, while preserving regional heterogeneity through multi-site sampling.
Systematic drug screening against seven clinically relevant agents revealed that organoid responses aligned with patient outcomes. In 14 relapsed patients, sensitivity or resistance to lenvatinib, sorafenib, or apatinib observed in organoids corresponded to clinical responses, demonstrating translational relevance. Integrative analyses identified multi-gene expression panels predictive of drug response, validated across training and validation cohorts.
Mechanistic work highlighted c-Jun as a key driver of lenvatinib resistance, acting through JNK and Wnt/β-catenin pathways. Knockdown of c-Jun resensitized resistant organoids, while a newly synthesized compound, PKUF-01, linking lenvatinib with a c-Jun inhibitor, showed synergistic efficacy both in vitro and in organoid-derived xenografts.
This study delivers a scalable organoid pharmacogenomics platform that not only captures the complexity of liver cancer heterogeneity but also enables discovery of predictive biomarkers and resistance mechanisms, paving the way for precision oncology in primary liver cancer.
A Pump‐Less Recirculating Organ-on-Chip for Islet–Liver Crosstalk
Type 2 diabetes, obesity, and metabolic dysfunction-associated steatotic liver disease (MASLD) are tightly interconnected conditions, yet studying their progression in humans remains challenging. Traditional animal models capture only parts of this complexity, and current in vitro systems often fail to reproduce the long-term, reciprocal signaling between key metabolic organs.
Aizenshtadt et al. (Advanced Healthcare Materials, 2024)(8) introduced a pump-less, dual recirculating organ-on-chip (rOoC) platform designed to model metabolic crosstalk between human pluripotent stem cell–derived islet and liver organoids. Unlike conventional pump-driven devices, this system relies on gravity-induced flow, making it more scalable and reducing artifacts such as unphysiological media-to-cell ratios.
The chip maintained the viability and function of both organoid types for at least two weeks. Islet organoids secreted insulin in response to glucose, while liver organoids consumed insulin and regulated glucose uptake, collectively sustaining euglycemia within physiological ranges. Under steatogenic conditions (fatty acids and fructose), liver organoids developed lipid accumulation and insulin resistance, while islets secreted pro-inflammatory cytokines, recapitulating early features of MASLD and type 2 diabetes. Importantly, treatment with known anti-diabetic drugs, including metformin and tolbutamide, reproduced established effects on glucose regulation and insulin responsiveness.
By demonstrating that a simplified, pump-free system can sustain organoid crosstalk and recapitulate both healthy and diseased metabolic states, this study highlights a practical route toward human-relevant models for drug discovery. Its ability to reproduce known pharmacological responses reinforces confidence that such liver–islet-on-chip systems may serve as predictive tools in the preclinical evaluation of metabolic therapies.
Cholangiocyte–Macrophage Crosstalk in the Biliary Niche via ORM2
Ductular reaction in chronic liver disease is characterized by expansion of biliary epithelial cells (BECs), macrophage accumulation, and fibrogenic activation, yet the epithelial signals orchestrating this niche have remained unclear.
Liu et al. (Gut, 2025)(9) identify the acute-phase protein orosomucoid 2 (ORM2) as a cholangiocyte-derived mediator that reprograms liver macrophages and reshapes the biliary microenvironment. Integrating transcriptome datasets from human and mouse livers with multiplex immunofluorescence, the authors show that ORM2 is upregulated in injured BECs but downregulated in injured hepatocytes, and that increased ORM2 signal in portal areas correlates with macrophage proximity.
Mechanistically, the study deploys multicellular organ-on-chip models: a biliary-niche-on-a-chip (BoC) and a perfused liver-on-a-chip (LoC) assembled from primary mouse liver and blood cells, complemented by patient-derived intrahepatic cholangiocyte organoids (hICOs) and human monocyte-derived macrophages. In these systems, cholangiocyte-derived ORM2 induces a distinct macrophage activation program with broad secretome changes and enhanced phagocytosis. In a complete LoC containing both hepatocytes and reactive BEC clusters, BEC-targeted Orm2 silencing reduced recruitment of Ly6C^high monocytes, indicating that cholangiocyte-origin ORM2 directs inflammatory cell trafficking in the portal niche.
The authors trace the mechanism to an ITPR2/CALM-dependent calcium pathway in macrophages: ORM2 elevates cytosolic Ca²⁺, upregulates calmodulins, and modulates lipid handling and cell-stress readouts; Itpr2 knockdown blunts these effects. Conditioned-medium experiments further show that ORM2-activated macrophages suppress cholangiocyte proliferation and increase apoptosis, with concordant responses in human hICOs from healthy and cirrhotic donors. Collectively, the work defines a cholangiocyte→macrophage→cholangiocyte signaling loop that promotes a pathogenic remodeling of the biliary niche, and illustrates how organoids and organ-on-chip can be combined to dissect multicellular mechanisms in human-relevant liver disease models.
UDPG-Driven Glycogenesis Suppresses Hepatic Lipogenesis
Excess conversion of postprandial glucose to fatty acids contributes to steatosis and progression to NAFLD. A central unresolved question is how hepatocytes prioritize glycogenesis versus lipogenesis when glucose influx rises.
Chen et al. (Science, 2025)(10) define a mechanism by which glycogenesis actively suppresses lipogenesis. Working with mouse and human primary hepatocytes and human liver organoids, the authors show that glucose carbon flows first into glycogen. Inhibiting glycogenic enzymes redirects flux toward lipid synthesis, indicating an antagonistic relationship between the two fates. Mechanistically, the glycogenesis intermediate uridine diphosphate glucose (UDPG) is transported to the Golgi by SLC35F5, where it binds site-1 protease (S1P) and prevents S1P-mediated cleavage of SREBP transcription factors. Using co-immunoprecipitation, click chemistry, LC-MS/MS, DARTS, molecular docking, and point mutagenesis, the study demonstrates direct UDPG–S1P interaction (requiring Asn438) and consequent S1P degradation via the ubiquitin–proteasome pathway. The result is diminished SREBP activation and down-regulation of lipogenic genes.
Functionally, exogenous UDPG promotes glycogenesis and reduces lipid accumulation in a mouse model of NAFLD and in human liver organoids, supporting translational relevance. These data position the UDPG–S1P–SREBP axis as a regulatory switch that biases hepatocytes toward glycogen storage while restraining de novo lipogenesis, with potential implications for therapeutic modulation of disordered hepatic fat metabolism.
References
- FDA. Roadmap to Reducing Animal Testing in Preclinical Safety Studies.
- Kong, L. R. et al. A glycolytic metabolite bypasses “two-hit” tumor suppression by BRCA2. Cell 187, 2269-2287.e16 (2024).
- Li, C. M.-C. et al. Brca1 haploinsufficiency promotes early tumor onset and epigenetic alterations in a mouse model of hereditary breast cancer. Nat. Genet. 56, 2763–2775 (2024).
- Nguyen, H. P. et al. Implantation of engineered adipocytes suppresses tumor progression in cancer models. Nat. Biotechnol. (2025) doi:10.1038/s41587-024-02551-2.
- Maulana, T. I. et al. Breast cancer-on-chip for patient-specific efficacy and safety testing of CAR-T cells. Cell Stem Cell 31, 989-1002.e9 (2024).
- Padrão, N. et al. TRIM24 as a therapeutic target in endocrine treatment–resistant breast cancer. Proc. Natl. Acad. Sci. 122, e2507571122 (2025).
- Lin, C.-J. et al. Genetic interactions reveal distinct biological and therapeutic implications in breast cancer. Cancer Cell 42, 701-719.e12 (2024).
- Li, Y. et al. Reprogramming intratumoral Treg cells by morpholino-mediated splicing of FOXP3 for cancer immunotherapy. Sci. Immunol. 10, eadr9933.
- Ascheid, D. et al. A vascularized breast cancer spheroid platform for the ranked evaluation of tumor microenvironment-targeted drugs by light sheet fluorescence microscopy. Nat. Commun. 15, (2024).
- Khalil, A. A. et al. A YAP-centered mechanotransduction loop drives collective breast cancer cell invasion. Nat. Commun. 15, 4866 (2024).
- Northey, J. J. et al. Mechanosensitive hormone signaling promotes mammary progenitor expansion and breast cancer risk. Cell Stem Cell 31, 106-126.e13 (2024).
FAQ
Drug discovery has been limited by a known trade-off. Primary human hepatocytes maintain adult functions but do not expand. Expandable cultures lose liver identity and drift into ductal metaplasia. This is not suitable for modelling metabolism or DILI. A 2025 paper reported a new human adult hepatocyte organoid system to address this issue. A specific blend of signaling molecules is used. This includes oncostatin M (OSM)–STAT3 activation. Long-term self-renewal is driven without flipping hepatocytes into a biliary fate. The organoids expand from single hepatocytes and maintain their hepatic identity. This platform is both expandable and adult-functional. It is a practical advancement for higher-fidelity liver organoid assays.
Metabolic zonation is the spatial division of labour across periportal (zone 1) and pericentral (zone 3) hepatocytes. This spatial arrangement is responsible for functions like nitrogen metabolism. It also determines zone-specific susceptibility to hepatotoxicants. This organisation is typically lost in conventional liver organoid systems. Multi-zonal human liver organoids (mZ-HLOs) were reported to be generated from pluripotent stem cells. Distinct zonal populations were created. Hepatic progenitors were engineered with inducible ascorbate synthesis for zone 1 identity. Low-dose bilirubin was applied to promote zone 3 fate. These populations self-assembled into fused organoids. Functionally, the organoids reproduced zone-specific responses to toxicants. Allyl alcohol selectively injured periportal regions. Acetaminophen affected pericentral zones.
The formation of a functional vasculature is a feature of early organogenesis. This process is inaccessible for direct study in human embryos. Existing organoid systems often lack a structured vascular network. This study combined micropatterned human pluripotent stem cells (hPSCs) with a screening of differentiation conditions. Gastruloid-based models with four lineage-specific fluorescent reporters were established. This allowed precise tracking of cell formation. A defined cocktail of growth factors was applied. Cardiac vascularized organoids (cVOs) were generated. These had a branched, lumenized vascular network integrated into multilineage cardiac tissue. This same strategy also produced hepatic vascularized organoids (hVOs). These hVOs contained hepatocytes interwoven with endothelial networks.
Cell-based therapies for liver failure are restricted. Restrictions are caused by the scarcity of donor hepatocytes and their rapid loss of function after transplantation. Encapsulated proliferating human hepatocyte organoids (eLOs) were reported as a potential therapeutic alternative. The method begins with proliferating human hepatocytes (ProliHHs). These were expanded in vitro and then matured into organoids. Encapsulation within alginate was performed. This provided structural support and immunoprotection. The organoids could then be transplanted intraperitoneally without host rejection. In mouse models of post-hepatectomy liver failure, eLOs restored liver functions. These functions included albumin production and ammonia detoxification. Treated animals showed improved survival. Safety assessments showed the organoids were genetically stable and non-tumorigenic.
Reproducing organ-specific vasculature is a challenge. In the liver, sinusoidal endothelial cells (LSECs) are needed for hepatocyte maturation. Most vascularized liver organoid systems lack sinusoidal features. A strategy was established to generate human liver bud organoids (HLBOs) with self-organised sinusoidal networks. Pluripotent stem cells were directed into liver sinusoidal endothelial progenitors (iLSEPs). These were then combined with hepatic endoderm, mesenchyme, and arterial progenitors. This combination was grown in a multilayered air–liquid interface (IMALI) culture. The resulting organoids contained hepatocyte clusters neighbored by multiple endothelial subtypes. The vasculature included LYVE1⁺STAB1⁺CD32b⁺ sinusoidal-like cells with fenestrae. WNT2-mediated angiocrine signaling from these cells enhanced hepatocyte differentiation.
The capacity for regeneration in the human liver during chronic injury has been uncertain. This study combined single-nucleus RNA sequencing (snRNA-seq) of 47 patient liver biopsies with 3D imaging. The biopsies covered the spectrum of metabolic dysfunction–associated steatotic liver disease (MASLD). Progressive architectural disruption was revealed. Hepatocytes lost zonation and the biliary tree was remodeled. Populations of cells emerged that co-expressed hepatocyte and cholangiocyte markers. These biphenotypic cells increased with disease severity. This indicates that epithelial plasticity is an acquired feature of chronic injury. Intrahepatic cholangiocyte organoids (ICOs) were generated from end-stage MASLD livers. These organoids recapitulated cholangiocyte-to-hepatocyte transdifferentiation. The PI3K–AKT–mTOR pathway was identified as a regulator of this process.
Inter- and intra-tumor heterogeneity is a challenge in liver cancer. It undermines the effectiveness of targeted therapies. A biobank of 399 patient-derived liver cancer organoids from 144 patients was established. These organoids faithfully recapitulated the histology, genomic landscape, and transcriptomic signatures of their parental tumors. Regional heterogeneity was also preserved. A systematic drug screening was performed. Organoid responses were found to align with patient outcomes. In 14 relapsed patients, sensitivity or resistance to lenvatinib or sorafenib in organoids corresponded to clinical responses. Mechanistic work highlighted c-Jun as a driver of lenvatinib resistance. This resistance acts through JNK and Wnt/β-catenin pathways. Knockdown of c-Jun resensitized resistant organoids.
Studying the progression of interconnected conditions like type 2 diabetes and MASLD is difficult. A pump-less, dual recirculating organ-on-chip (rOoC) platform was introduced. It was designed to model metabolic crosstalk between human pluripotent stem cell–derived islet and liver organoids. This system relies on gravity-induced flow. This makes it more scalable. It also reduces artifacts, such as unphysiological media-to-cell ratios, found in pump-driven devices. The chip maintained the viability and function of both organoid types for at least two weeks. Islet organoids secreted insulin, while liver organoids consumed it. Under steatogenic conditions, liver organoids developed lipid accumulation and insulin resistance. Islets secreted pro-inflammatory cytokines. This recapitulated early features of MASLD and type 2 diabetes.
The epithelial signals orchestrating the ductular reaction in chronic liver disease have been unclear. The acute-phase protein orosomucoid 2 (ORM2) was identified as a cholangiocyte-derived mediator. This protein reprograms liver macrophages. ORM2 is upregulated in injured biliary epithelial cells (BECs). Multicellular organ-on-chip models were used. These included a biliary-niche-on-a-chip (BoC) and a perfused liver-on-a-chip (LoC). Patient-derived intrahepatic cholangiocyte organoids (hICOs) were also used. In these systems, cholangiocyte-derived ORM2 was shown to induce a distinct macrophage activation program. A cholangiocyte→macrophage→cholangiocyte signaling loop was defined. This loop promotes a pathogenic remodeling of the biliary niche. The work illustrates how organoids and organ-on-chip can be combined.
A central question is how hepatocytes prioritize glycogenesis (glycogen synthesis) versus lipogenesis (fat synthesis) when glucose increases. A mechanism by which glycogenesis actively suppresses lipogenesis was defined. Mouse and human primary hepatocytes and human liver organoids were used. It was shown that glucose carbon flows first into glycogen. The glycogenesis intermediate uridine diphosphate glucose (UDPG) is transported to the Golgi by SLC35F5. There, it binds site-1 protease (S1P). This binding prevents S1P-mediated cleavage of SREBP transcription factors. This interaction was demonstrated with co-immunoprecipitation and molecular docking. The result is diminished SREBP activation. Lipogenic genes are consequently down-regulated. Exogenous UDPG was shown to reduce lipid accumulation in human liver organoids.