Breast cancer remains the most common malignancy in women worldwide, and progress in treatment increasingly depends on models that more faithfully recapitulate human disease. Over the past two years, major advances have been made through the adoption of human-derived in vitro systems such as patient-derived organoids and microphysiological platforms (organ-on-chip). These technologies capture tumor heterogeneity, microenvironmental cues, and therapy responses with a fidelity that surpasses traditional 2D cultures or animal models, while aligning with 3Rs principles of reducing animal use.
Researchers are now integrating these in vitro models with traditional in vivo data and with computational approaches, creating a more comprehensive and predictive research framework. This strategy is not only being adopted in academic laboratories but also encouraged at the regulatory and industrial level. This was exemplified by the FDA and NIH that have explicitly promoted such integrative methodologies as part of their roadmap to reduce reliance on animal testing, highlighting organoids, organ-on-chip systems, and computational modeling as validated tools to accelerate translation and improve patient safety(1).
In this article, we highlight 10 key studies on breast cancer research published in the last two years. Each demonstrates how innovative human-derived in vitro systems (sometimes combined with in vivo or computational approaches) are driving breast cancer research and paving the way toward more precise and patient-centered therapies.
Learn more about our ready to use breast cancer organoid models.

A Glycolytic Byproduct That Disarms BRCA2
For decades, the “two-hit hypothesis” has guided our understanding of tumor suppressor genes like BRCA2: both alleles must be inactivated to unleash cancer-promoting genomic instability. Yet a recent study by Kong et al. (Cell, 2024)(2) shows this rule can be bypassed by a surprising culprit: the glycolytic byproduct methylglyoxal (MGO).
MGO accumulates when glycolysis runs at high flux, as in cancer, diabetes, or inflammatory states. Kong and colleagues discovered that MGO induces proteolysis of BRCA2, temporarily impairing its DNA repair function. In mammary epithelial cells and patient-derived breast organoids carrying only one mutant BRCA2 allele, brief MGO exposure produced the characteristic mutational “scar” of BRCA2 deficiency — despite the second allele being intact. This reveals a mechanism of functional haploinsufficiency, where BRCA2 activity falls below the threshold needed for genome stability.
The consequences are profound. Instead of requiring a genetic “second hit,” episodic bursts of MGO-driven BRCA2 loss can seed mutations that fuel tumor evolution. This connects metabolic state to cancer risk: oncogene-driven glycolysis, diet, or metabolic disease may influence breast cancer initiation in BRCA2 carriers. By linking cell metabolism, DNA repair, and genome mutagenesis, this work reshapes how we think about early tumorigenesis and suggests new avenues for prevention.
BRCA1 Haploinsufficiency Leaves Epigenetic Scars that Accelerate Tumor Onset
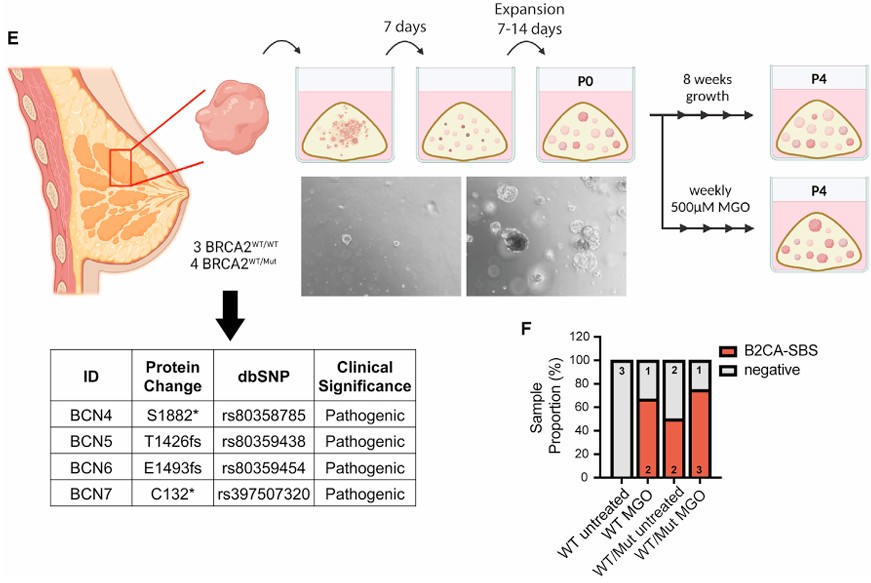
The traditional “two-hit” model holds that both alleles of a tumor suppressor must be lost for cancer to emerge. Yet, as shown by Kong et al. in early 2024 with BRCA2 and the glycolytic metabolite methylglyoxal, this rule is not absolute. Later this year, Li et al. (Nature Genetics, 2024)(3) showed that even a single defective copy of BRCA1 can prime breast epithelial cells for malignancy.
Using engineered mouse models, the team compared animals born heterozygous for Brca1 with those in which Brca1 was deleted later in life. Tumors arose much earlier in the heterozygous mice, suggesting that BRCA1 haploinsufficiency accelerates tumorigenesis. Single-cell RNA-seq revealed minimal transcriptional differences, but chromatin accessibility profiling uncovered widespread epigenetic scars: loss of differentiation-linked ELF5 sites and gain of AP-1/JUN motifs activating Wnt10a signaling.
To test function, the authors generated mammary organoids from heterozygous cells. These 3D cultures recapitulated early tissue structure and showed that Wnt10a activation drove abnormal proliferation and disorganized growth, hallmarks of pre-neoplastic change. The organoid system provided direct experimental evidence that early chromatin changes translate into altered behavior.
Together, Li et al. and Kong et al. provide solid evidence and mechanistic insights that refine our understanding of the ‘two-hit’ hypothesis and tumorigenesis, offering a more nuanced view that also raises new questions about the role of metabolic disorders, glycolytic reprogramming, and epigenetic priming in shaping early cancer risk.
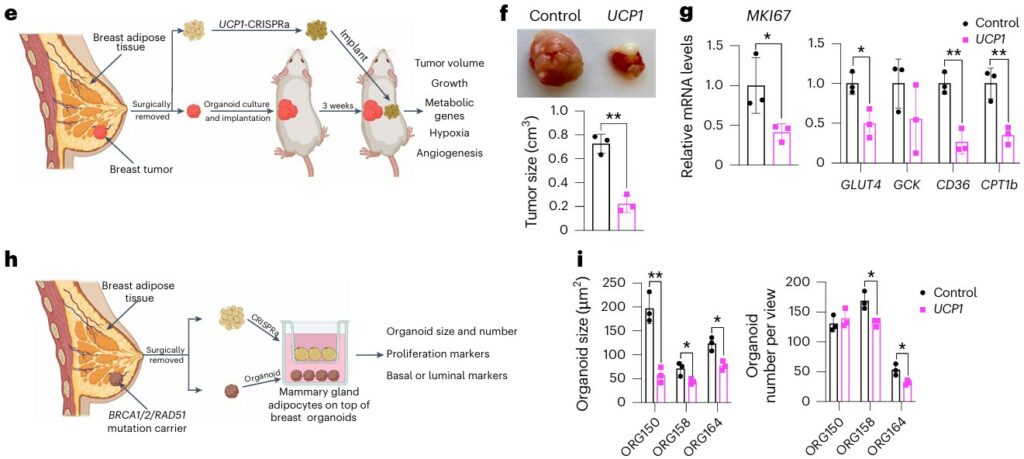
Engineered Adipocytes Outcompete Tumors for Fuel
his study is particularly striking for the simplicity of its concept: To starve the tumor by feeding another cell type. On the other hand, it required cutting-edge bioengineering to realize. We covered it earlier this year in our Organoids Digest (February issue), and its impact has only grown since.
Nguyen et al. (Nature Biotechnology, 2025)(4) developed a therapeutic approach they call adipose manipulation transplantation (AMT). By using CRISPR activation to upregulate genes such as UCP1, PPARGC1A, or PRDM16, the team converted human white adipocytes into highly metabolically active, “brown-like” fat cells (beige adipocytes). These engineered adipocytes consume large amounts of glucose and fatty acids, effectively outcompeting tumors for nutrients.
When co-cultured with breast cancer organoids, the engineered fat cells suppressed cancer proliferation, lowering glycolysis and fatty acid oxidation in tumor cells. Implanted as adipose tissue organoids alongside xenografts of breast, pancreatic, or prostate cancer, they reduced tumor growth, angiogenesis, and hypoxia. Remarkably, the strategy worked even in genetic mouse models of breast cancer, whether the engineered organoids were placed adjacent to or distant from tumors.
Importantly, the therapy is controllable and customizable: activation can be toggled with tetracycline, and nutrient competition tailored by targeting different metabolic pathways. This elegant approach reframes the tumor microenvironment as a metabolic battleground, and suggests that beiging strategy with adipocytes may serve as living therapeutics against cancer.
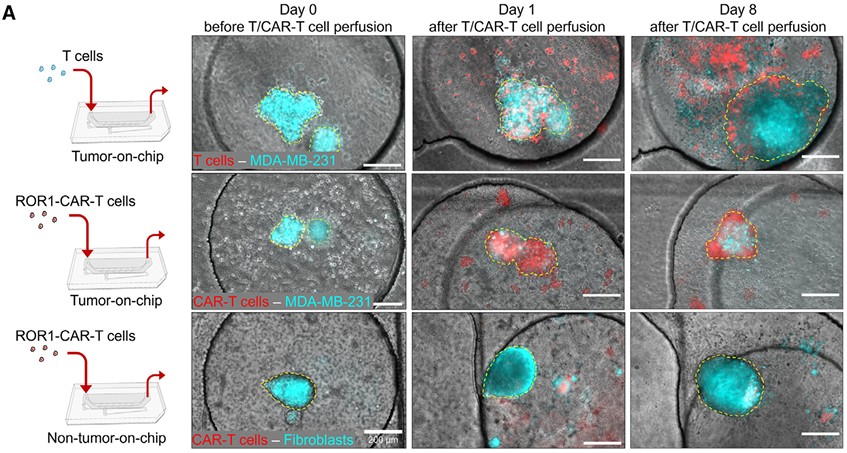
Breast Cancer-on-Chip Brings CAR-T Testing Closer to Patients
CAR-T therapy has transformed hematological cancers, but its translation to solid tumors like breast cancer has been slow, hampered by antigen heterogeneity, stromal barriers, and risks such as cytokine release syndrome (CRS). In a paper recognized among the Best of Cell Stem Cell 2024, Maulana et al.(5) present an elegant solution: a breast cancer-on-chip platform for patient-specific testing of CAR-T efficacy and safety.
The system integrates patient-derived breast cancer organoids with a perfused, endothelialized microchannel that mimics the tumor vasculature. CAR-T cells infused through the channel must traverse the endothelial barrier before engaging tumor cells, recapitulating a key clinical hurdle. The platform enables long-term monitoring of CAR-T infiltration, tumor killing, and cytokine dynamics.
In this system, CAR-T cells eliminated tumor organoids in an antigen-specific manner and generated cytokine release profiles that mirrored clinical CRS. Importantly, the chip reproduced patient-specific responses, with CAR-T efficacy scaling to ROR1 antigen density in organoids. The system also provided a preclinical model for safety switches, showing that dasatinib could transiently suppress CAR-T activity to control cytokine toxicity without abolishing efficacy.
By combining organoids, microfluidics, and immune cells, this study delivered a scalable precision-oncology tool that could de-risk CAR-T therapies and accelerate their entry into solid tumor care.
Targeting TRIM24 to Overcome Endocrine Resistance in Breast Cancer
Around 70% of breast cancers are estrogen receptor alpha (ERα)-positive, meaning they rely on estrogen signaling for growth and survival. When activated, ERα binds DNA, recruits cofactors, and switches on genes that drive cell-cycle progression, survival pathways, and metastatic programs. This dependency exemplifies transcriptional addiction (a state where tumors become reliant on hyperactive transcriptional programs and their supporting machinery for survival). Endocrine therapies like tamoxifen or aromatase inhibitors aim to block this pathway, but roughly one-third of patients relapse due to ESR1 mutations or adaptive use of cofactors that sustain ERα activity.
In Padrão et al. (PNAS, 2025)(6), researchers identify the chromatin reader TRIM24 as a pivotal ERα cofactor. TRIM24 maintains active histone marks and stabilizes ERα binding across the genome. Using a heterobifunctional degrader, dTRIM24, they selectively eliminated TRIM24, suppressing ERα-driven transcription and blocking proliferation in both endocrine-sensitive and resistant models, including ESR1-mutant lines. In patient-derived breast cancer organoids, dTRIM24 sharply reduced viability of ERα+ tumors, while sparing triple-negative controls.
The findings refined the concept of transcriptional addiction: ERα+ tumors are not only addicted to ERα itself, but also to the chromatin machinery that sustains its activity. Targeting cofactors like TRIM24 could therefore bypass resistance mechanisms and extend therapy effectiveness in metastatic disease.
Genetic Interactions Redraw the Map of Breast Cancer Therapy
Precision oncology often treats single driver mutations as the key to therapy choice. But cancers rarely operate through isolated alterations. In a landmark study published in Cancer Cell (2024), Lin et al.(7) charted a comprehensive map of genetic interactions in breast cancer, revealing how combinations of mutations shape biology and therapeutic response in ways single mutations cannot capture.
The team analyzed two large cohorts from Fudan University; 873 patients with multi-omics profiling and 4,405 patients with genomic data linked to real-world clinical outcomes, validated with TCGA, METABRIC, and other datasets. They built a network of co-occurring and mutually exclusive alterations, then used patient-derived organoids, tumor fragments, mini-PDX, and xenografts to test functional consequences.
Key discoveries included: TP53 mutations plus AURKA amplification drove endocrine resistance, confirmed in organoids and mini-PDX; germline BRCA1 loss plus MYC amplification enhanced PARP inhibitor sensitivity; and TP53 mutation plus MYB amplification induced immune evasion in TNBC organoid–T cell co-cultures. These interactions provided far stronger predictions of therapy response than single drivers alone.
By integrating genomics with functional organoid validation, this work reframes precision oncology as a network-based discipline. It highlights that effective therapy choices will increasingly depend on co-alteration profiles, rather than single mutation.
Reprogramming Treg Cells by Targeting FOXP3 Isoforms
Regulatory T cells (Tregs) are a major obstacle to effective antitumor immunity, as they suppress cytotoxic T cell activity within the tumor microenvironment. In a striking study, Li et al.(8) revealed that the isoform balance of the master Treg regulator FOXP3 can decisively alter this immune suppression. Humans express two major FOXP3 variants: the full-length form (FOXP3FL) and a shorter isoform lacking exon 2 (FOXP3dE2).
Using a mouse model engineered to express only FOXP3dE2, the authors showed remarkable resistance to tumor development across multiple cancer types. Treg cells in these mice were less suppressive, instead producing effector cytokines such as IFN-γ, which boosted CD8 T cell responses. Importantly, analysis of human data revealed that FOXP3dE2 expression in triple-negative breast cancer correlated with improved survival, underscoring clinical relevance.
To translate this insight into therapy, the team designed a morpholino oligo that induced skipping of exon 2, shifting expression toward FOXP3dE2. In mouse tumor models, and in assays using patient-derived breast and colorectal cancer organoids, this strategy enhanced CD8 T cell–mediated tumor killing.
By demonstrating that Treg cell reprogramming through FOXP3 isoform modulation can complement existing immune checkpoint inhibitors and unleash antitumor immunity, this work opens a new therapeutic avenue for solid tumors, including breast cancer.
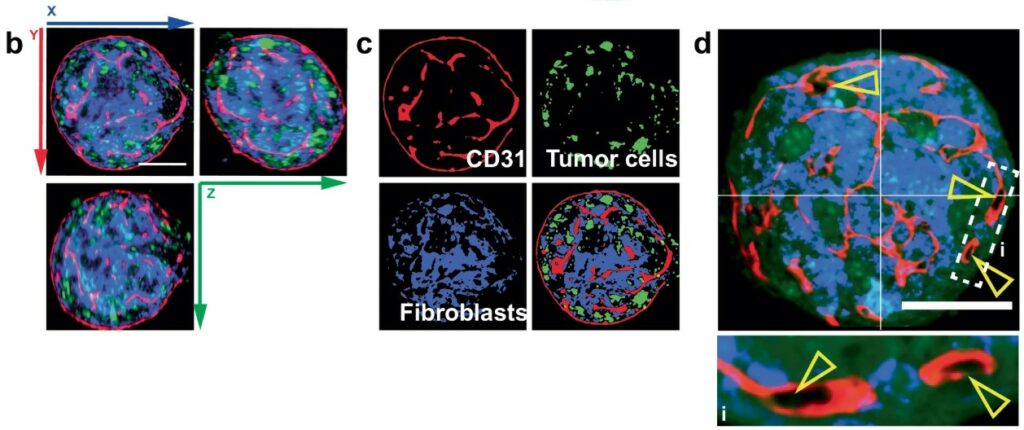
Vascularized Breast Cancer Spheroids to Study Tumor Microenvironment-Targeting Drugs
Targeting the tumor microenvironment (TME), including fibroblasts, vasculature, immune infiltrates, and extracellular matrix, remains one of the most difficult challenges in cancer therapy. Yet most preclinical assays cannot capture these intricate interactions. In Nature Communications (2024), Ascheid et al.(9) present a breakthrough platform: vascularized breast cancer spheroids (VTSs) that self-organize from tumor cells, fibroblasts, endothelial cells, and macrophages, all of human origin. These spheroids develop complex architectures, including pseudovasculature networks, making them far more representative of breast tumors than traditional spheroids.
The system is modular, reproducible, and adaptable to different breast cancer subtypes or genetic manipulations. Crucially, it was engineered for compatibility with standard biotech industry and academic laboratory settings; using 96-well plates, primary cells commercially available, and imaging via light sheet fluorescence microscopy with automated analysis pipelines. This makes high-content screening of TME-targeted therapies feasible beyond specialized organ-on-chip labs.
Using the platform, the authors ranked anti-angiogenic and antifibrotic drug candidates, capturing subtle but clinically relevant differences in how they remodel vessels, fibroblasts, and immune infiltration. Even closely related compounds showed divergent effects depending on the tumor model.
By combining scalability with physiological relevance, vascularized tumor spheroids provide a robust bridge between reductionist 2D assays and complex organ-on-chip systems, enabling more predictive drug development for TME-targeted therapies in breast cancer.
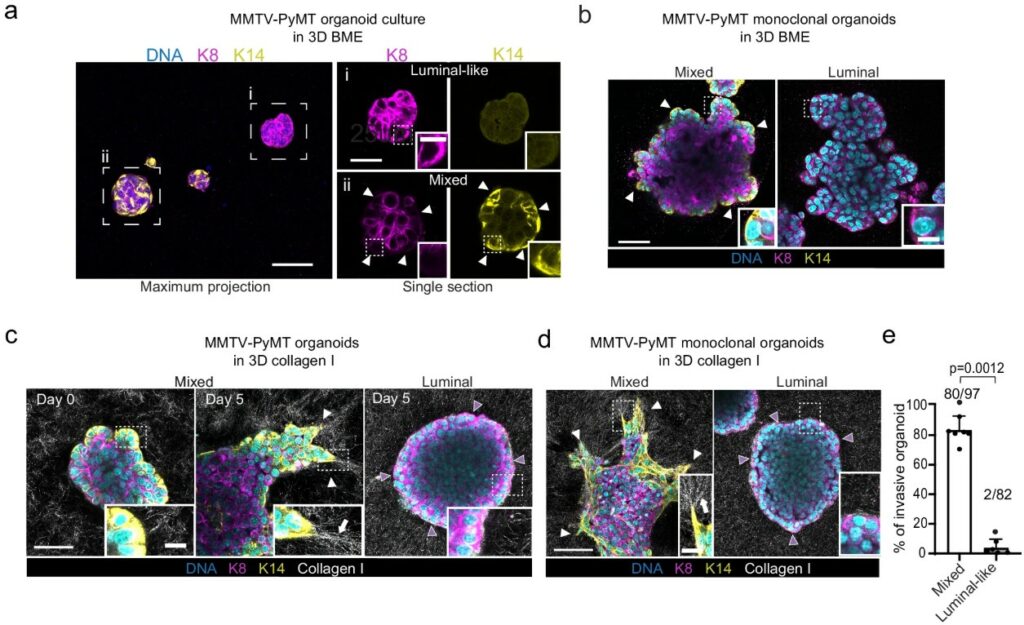
The Mechanism of Leader Cell Emergence
Collective cancer invasion is driven by leader cells, a specialized subset of tumor cells at the tumor-ECM interface that spearhead the process by remodeling the matrix to create paths for follower cells. Khalil et al.(10) elucidated a YAP-centered mechanotransduction feed-forward loop as a key driver for the emergence of these cells. The authors showed that contact with a Collagen I matrix activates the transcriptional coactivator YAP in specific “basal-like” cells. This initiates a transcriptional program for ECM remodeling that increases mechanical tension in the collagen fibers. This amplified tension is then transduced back to the cells, causing hyper-activation of YAP and solidifying the invasive leader cell phenotype.
This self-reinforcing, tension-dependent mechanism was confirmed by its abrogation in floating collagen gels, where mechanical forces cannot be sustained. Functional inhibition of the YAP-TEAD axis via shRNA or small molecules significantly reduced invasion in mouse and patient-derived organoids.
Critically, the study utilized CRISPR/Cas9 to show that this invasive program is independent of Keratin 14 (K14) expression, redefining its role as a marker rather than a functional driver. This work provides a new paradigm for cancer invasion, highlighting a therapeutically targetable mechanical interplay between tumor cells and their microenvironment.
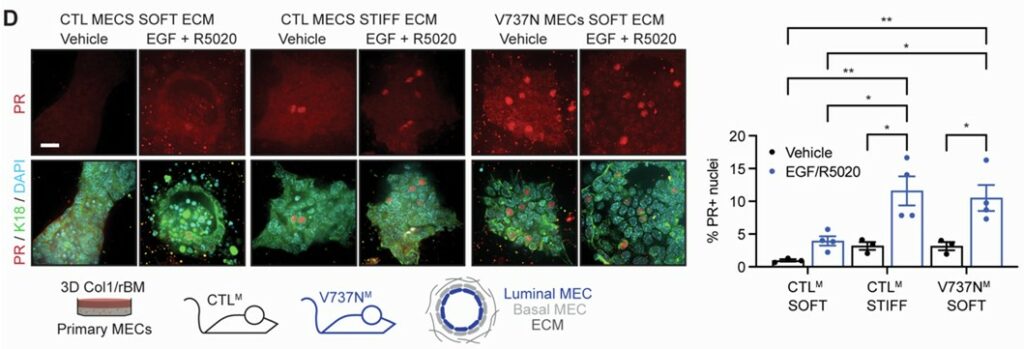
A Mechanical Link Between Breast Density and Cancer Risk
High mammographic density, characterized by stiff, collagen-rich breast tissue, is a major risk factor for breast cancer. A study by Northey et al.(11) provides a direct molecular explanation for this link, using clinical samples, genetically engineered mice, and 3D organoid cultures to show how mechanical forces amplify hormone signaling to expand the pool of cancer-prone mammary progenitor cells.
The mechanism begins when a stiff extracellular matrix is sensed by integrin receptors, activating the enzyme ERK. This in turn hyper-activates the progesterone receptor (PR), driving the production of the signaling protein RANKL, which stimulates progenitor cell proliferation.
This was tested directly using mammary organoids cultured in hydrogels of varying stiffness. Organoids grown in a “stiff” environment showed significantly elevated RANKL production in response to hormones, an effect that was blocked by inhibiting key mechanosignaling enzymes. The authors validated this full pathway in mouse models, demonstrating that inhibiting either ERK or RANKL signaling prevents both progenitor expansion and the formation of early tumor lesions.
The importance of this study lies into how it unifies tissue mechanics and hormone biology and identifies the RANKL pathway as a promising target for breast cancer prevention.
References
- FDA. Roadmap to Reducing Animal Testing in Preclinical Safety Studies.
- Kong, L. R. et al. A glycolytic metabolite bypasses “two-hit” tumor suppression by BRCA2. Cell 187, 2269-2287.e16 (2024).
- Li, C. M.-C. et al. Brca1 haploinsufficiency promotes early tumor onset and epigenetic alterations in a mouse model of hereditary breast cancer. Nat. Genet. 56, 2763–2775 (2024).
- Nguyen, H. P. et al. Implantation of engineered adipocytes suppresses tumor progression in cancer models. Nat. Biotechnol. (2025) doi:10.1038/s41587-024-02551-2.
- Maulana, T. I. et al. Breast cancer-on-chip for patient-specific efficacy and safety testing of CAR-T cells. Cell Stem Cell 31, 989-1002.e9 (2024).
- Padrão, N. et al. TRIM24 as a therapeutic target in endocrine treatment–resistant breast cancer. Proc. Natl. Acad. Sci. 122, e2507571122 (2025).
- Lin, C.-J. et al. Genetic interactions reveal distinct biological and therapeutic implications in breast cancer. Cancer Cell 42, 701-719.e12 (2024).
- Li, Y. et al. Reprogramming intratumoral Treg cells by morpholino-mediated splicing of FOXP3 for cancer immunotherapy. Sci. Immunol. 10, eadr9933.
- Ascheid, D. et al. A vascularized breast cancer spheroid platform for the ranked evaluation of tumor microenvironment-targeted drugs by light sheet fluorescence microscopy. Nat. Commun. 15, (2024).
- Khalil, A. A. et al. A YAP-centered mechanotransduction loop drives collective breast cancer cell invasion. Nat. Commun. 15, 4866 (2024).
- Northey, J. J. et al. Mechanosensitive hormone signaling promotes mammary progenitor expansion and breast cancer risk. Cell Stem Cell 31, 106-126.e13 (2024).
FAQ
Progress in breast cancer treatment relies on models that more faithfully show human disease. Human-derived in vitro systems are being adopted. These technologies surpass traditional 2D cultures or animal models in fidelity. Tumour heterogeneity, microenvironmental cues, and therapy responses are captured by these platforms. The reduction of animal use is also supported by this approach. A more complete research framework is created by integrating these in vitro models with computational approaches. This strategy is encouraged at the regulatory level. The FDA and NIH have encouraged these methodologies. This is part of a plan to reduce animal testing.
The “two-hit hypothesis” for tumour suppressors like BRCA2 has been re-examined. A glycolytic byproduct, methylglyoxal (MGO), was found to be involved. MGO accumulates when glycolysis runs at a high flux. It was discovered that MGO induces the proteolysis (breakdown) of BRCA2. This action temporarily impairs its DNA repair function. Mammary epithelial cells and patient-derived breast organoids were used. These cells carried only one mutant BRCA2 allele. Brief MGO exposure was shown to produce the characteristic mutational “scar” of BRCA2 deficiency. This occurred even though the second allele was intact. A mechanism of functional haploinsufficiency is revealed. A genetic “second hit” is not required. Episodic bursts of MGO-driven BRCA2 loss can seed mutations. This work connects metabolic state to cancer risk.
The traditional “two-hit” model has been further examined. It was shown that even a single defective copy of BRCA1 can prime breast epithelial cells for malignancy. This state is called haploinsufficiency. Tumours were observed to arise much earlier in mice heterozygous for Brca1. This suggested that haploinsufficiency accelerates tumorigenesis. Widespread epigenetic scars were uncovered by chromatin accessibility profiling. These included the loss of differentiation-linked ELF5 sites. A gain of AP-1/JUN motifs was also found, which activated Wnt10a signaling. Mammary organoids were generated from heterozygous cells. These 3D cultures were used to test the function. It was shown that Wnt10a activation drove abnormal proliferation. Disorganized growth, a hallmark of pre-neoplastic change, was also observed. Experimental evidence was provided by the organoid system.
A therapeutic approach called adipose manipulation transplantation (AMT) was developed. CRISPR activation was used. Genes such as UCP1, PPARGC1A, or PRDM16 were upregulated. This process converted human white adipocytes into highly metabolically active, “beige” adipocytes. These engineered adipocytes consume large amounts of glucose and fatty acids. Tumours are effectively outcompeted for nutrients. When these engineered cells were co-cultured with breast cancer organoids, cancer proliferation was suppressed. Lowered glycolysis and fatty acid oxidation in tumour cells were observed. The cells were also implanted as adipose tissue organoids alongside xenografts. Tumour growth, angiogenesis, and hypoxia were reduced. The strategy was also shown to work in genetic mouse models of breast cancer.
The translation of CAR-T therapy to solid tumours like breast cancer has been slow. A breast cancer-on-chip platform for patient-specific testing of CAR-T efficacy and safety was presented. Patient-derived breast cancer organoids are integrated by the system. A perfused, endothelialized microchannel that copies the tumour vasculature is also included. CAR-T cells are infused through the channel. They must traverse the endothelial barrier before engaging tumour cells. This process recapitulates a clinical hurdle. Long-term monitoring of CAR-T infiltration, tumour killing, and cytokine dynamics is enabled by the platform. CAR-T cells were shown to eliminate tumour organoids in an antigen-specific manner. Cytokine release profiles that mirrored clinical CRS were also generated. Patient-specific responses were reproduced.
Approximately 70% of breast cancers are estrogen receptor alpha (ERα)-positive. They rely on estrogen signaling for growth. Endocrine therapies block this pathway. About one-third of patients relapse due to ESR1 mutations or the use of cofactors that sustain ERα activity. The chromatin reader TRIM24 was identified by researchers as an ERα cofactor. TRIM24 maintains active histone marks. It also stabilizes ERα binding across the genome. A heterobifunctional degrader, dTRIM24, was used. TRIM24 was selectively eliminated by this degrader. This suppressed ERα-driven transcription. Proliferation was blocked in both endocrine-sensitive and resistant models, including ESR1-mutant lines. In patient-derived breast cancer organoids, dTRIM24 sharply reduced viability. This was seen in ERα+ tumours, while triple-negative controls were spared.
Precision oncology often treats single driver mutations as the guide for therapy choice. Cancers rarely operate through isolated alterations. A map of genetic interactions in breast cancer was charted. This map revealed how combinations of mutations shape biology. Therapeutic response is also shaped in ways single mutations cannot capture. Two large patient cohorts were analysed. Patient-derived organoids, tumour fragments, and xenografts were used to test functional consequences. One discovery was that TP53 mutations plus AURKA amplification drove endocrine resistance. This was confirmed in organoids. Another finding was that germline BRCA1 loss plus MYC amplification increased PARP inhibitor sensitivity. TP53 mutation plus MYB amplification induced immune evasion in TNBC organoid–T cell co-cultures. These co-alteration profiles provided much stronger predictions of therapy response.
Targeting the tumour microenvironment (TME) remains a difficult challenge. Most preclinical assays cannot capture these interactions. A platform of vascularized breast cancer spheroids (VTSs) was presented. These spheroids self-organise. They are formed from tumour cells, fibroblasts, endothelial cells, and macrophages, all of human origin. Detailed architectures are developed by these spheroids. These architectures include pseudovasculature networks. This makes them more representative of breast tumours than traditional spheroids. The system is modular and reproducible. It is also adaptable to different breast cancer subtypes. It was engineered for compatibility with standard 96-well plates. This makes high-content screening of TME-targeted therapies feasible. Anti-angiogenic and antifibrotic drug candidates were ranked using the platform.
Collective cancer invasion is headed by leader cells. These are a specialized subset of tumour cells. They remodel the matrix to create paths for follower cells. A YAP-centered mechanotransduction feed-forward loop was identified as a mechanism for the emergence of these cells. It was shown that contact with a Collagen I matrix activates the transcriptional coactivator YAP in specific cells. A transcriptional program for ECM remodeling is initiated. This action increases mechanical tension in the collagen fibres. This amplified tension is then transduced back to the cells. Hyper-activation of YAP is caused, and the invasive leader cell phenotype is solidified. This self-reinforcing mechanism was confirmed. Functional inhibition of the YAP-TEAD axis reduced invasion in mouse and patient-derived organoids.
High mammographic density is a known risk factor for breast cancer. This condition is characterized by stiff, collagen-rich breast tissue. A direct molecular explanation for this link was provided. Mechanical forces increase hormone signaling. This increase expands the pool of cancer-prone mammary progenitor cells. The mechanism begins when a stiff extracellular matrix is sensed by integrin receptors. This action activates the enzyme ERK. The progesterone receptor (PR) is then hyper-activated. This leads to the production of the signaling protein RANKL. Progenitor cell proliferation is stimulated by RANKL. This was tested directly using mammary organoids cultured in hydrogels of varying stiffness. Organoids grown in a “stiff” environment showed elevated RANKL production in response to hormones. This effect was blocked by inhibiting mechanosignaling enzymes.